Hervé Falet, PhD
Investigator
Hematopoiesis and Immunology

Associate Professor
Department of Cell Biology, Neurobiology and Anatomy
Medical College of Wisconsin
Education and Training
Postdoctoral: Medicine, Brigham and Women's Hospital and Harvard Medical School
PhD: Molecular and Cellular Biology, Paris Descartes University
MS: Molecular Pharmacology, Paris Descartes University
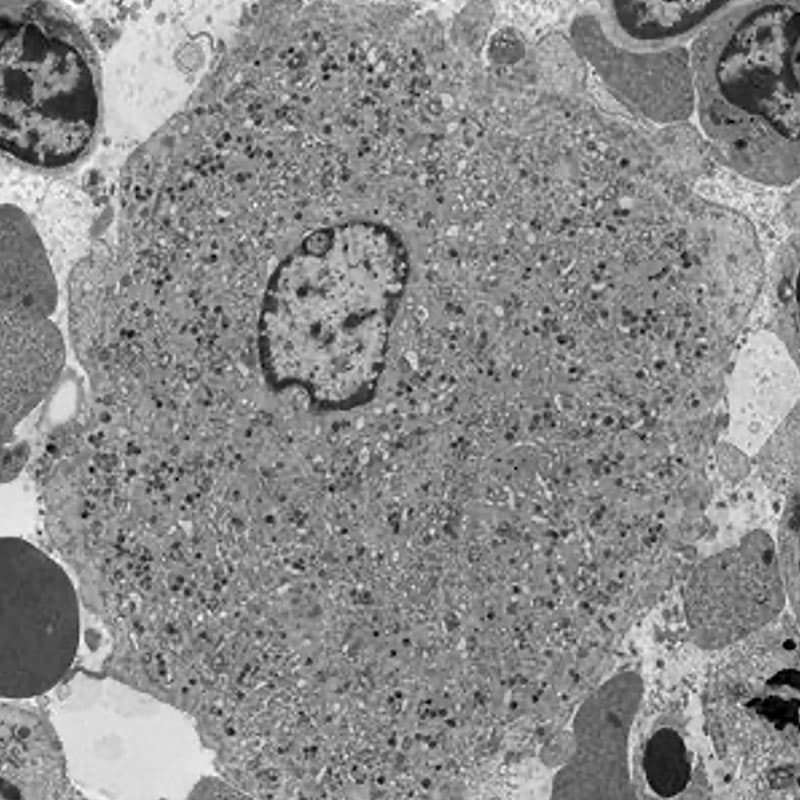
Fig.1. Transmission electron microscopy of a mouse bone marrow megakaryocyte.
Platelet Production
Blood platelets are produced in the bone marrow by megakaryocytes in unique processes that require polyploidisation and extensive membrane rearrangements. These include the formation of the demarcation membrane system, the surface-connected membrane reservoir for future platelets. As megakaryocytes mature, the demarcation membrane system initiates as a single plasma membrane invagination that extends between the lobes of the polyploid nucleus and further expands into its mature form by addition of intracellular membrane materials that are biosynthesized in the endoplasmic reticulum or transported from the Golgi apparatus. The precise molecular mechanisms responsible for these unique membrane rearrangements remain poorly understood. Our current research is focused on the role of receptor-mediated endocytosis in platelet production and function, focusing on the F-BAR protein PACSIN2 and the large GTPase dynamin 2.

Fig.2. α-tubulin (green) and GPIbα (red) immunofluorescence images of a mouse fetal liver–derived megakaryocyte forming proplatelets in vitro.
PACSIN2
PACSIN2 belongs to the F-BAR family of proteins that bind lipid bilayers to generate membrane tubular invaginations in cells. We have identified PACSIN2 as an internal component of the initiating demarcation membrane system in megakaryocytes, where its membrane tabulation activity is regulated by the cytoskeletal and scaffolding protein filamin A (Begonja, Pluthero et al. Blood. 2015;126(1):80-88), a critical regulator of platelet production and function (Falet et al. J Exp Med. 2010;107(9):1967-1979; Begonja et al. Blood. 2011;118(8):2285-2295). Single nucleotide variants in PACSIN2 have recently been associated with altered platelet count and mean platelet volume. We have obtained Pacsin2–/– mice from Dr Markus Plomann (University of Cologne, Germany) to further investigate the role of PACSIN2 in platelet production and function. Pacsin2–/– mice develop mild thrombocytopenia (Biswas, Boyd et al. J Thromb Haemost. 2023 [Epub ahead of print]).
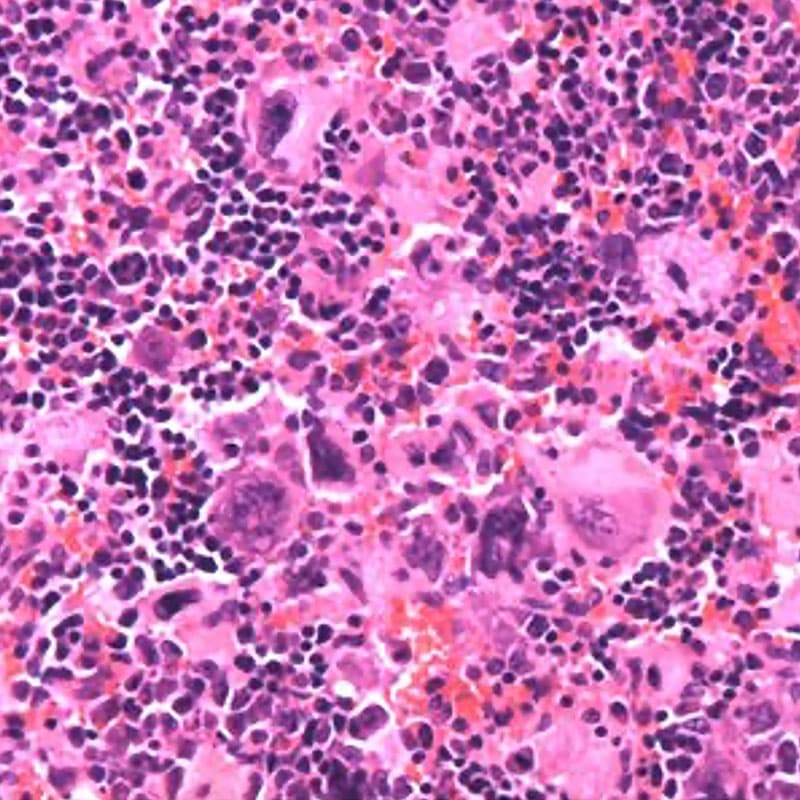
Fig.3. Hematoxylin and eosin staining of Dnm2Plt–/– spleens displaying megakaryocyte hyperplasia.
Myelofibrosis
Dnm2Plt–/– mice also develop hallmarks of myelofibrosis, such as massive megakaryocyte hyperplasia, hematopoietic stem and progenitor cell proliferation, bone marrow fibrosis, extramedullary hematopoiesis, and severe splenomegaly (Bender, Giannini et al. Blood. 2015;125(6):1014-1024). The phenotype is reminiscent of patients with myelofibrosis that is characterized by uncontrolled JAK/STAT signaling in hematopoietic stem cells. At the cellular level, Dnm2Plt–/– platelets display constitutive activation but decreased expression of the tyrosine kinase JAK2 and are unable to endocytose the thrombopoietin receptor Mpl, leading to elevated circulating thrombopoietin levels (Eaton et al. Front Oncol. 2022;12:959806).
In summary, our research addresses the gap of knowledge on receptor-mediated endocytosis in platelet production and function. We anticipate that findings obtained from these studies will yield better understanding to obtain basic science information on how membrane rearrangements contribute to platelet production and function, and to develop treatments for effective re-establishment of platelet production in the setting of thrombocytopenia.