Robert Montgomery, MD

Over the past year we have 1) demonstrated that FVIII is made in endothelial cells; 2) completed enrollment of our historical type 1 Von Willebrand disease cohort of 500 families done across the USA; 3) initiated a prospective study of newly diagnosed VWD patients across the USA; 4) did BM TxP with gene therapy of the stem cells to direct FVIII production in platelets; and 5) initialed studies to study acquired abnormalities of von Willebrand factor in newborns having open heart surgery for complex congenital heart disease.
While scientists have recognized for years that liver transplant can cure hemophilia and concluded that FVIII must be made in the liver hepatocyte. Using various mouse models that we developed, studies were possible to block the FVIII production in a single type of cell. For example we can remove the FVIII functional gene from all hepatocytes. Interestingly, the FVIII level in the mouse plasma was normal and not affected – thereby proving that hepatocytes didn’t make FVIII. We then did further “tissue-specific” knockout or elimination of the functional FVIII gene from other cells and thereby proved that if you stopped FVIII production in endothelial cells, there was no FVIII in plasma and thereby proved that FVIII was produced in endothelial cells where it was stored and released physiologically with its companion molecule von Willebrand factor.
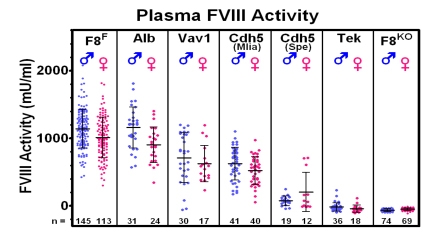

This figure demonstrates that crossing our mouse with a “floxed” FVIII gene with mice with tissue-specific “Cre-recombinase” demonstrate no reduction of FVIII in the albumin-Cre (hepatocyte-specific) mouse, but total elimination of plasma FVIII in the Tek-Cre (endothelial cell specific) mouse.
We have finished the 7th year of a multinational program project that is focused on von Willebrand Disease (VWD). VWD is probably the most common hereditary bleeding disorder and may affect 1 in 50 to 1 in 1000 of our general population. In studying our cohort of 500 families with the common form of VWD, type 1 VWD, we found that not all of those subjects continue to have abnormal levels of VWF. We do find that most have a gene mutation in their VWF gene that can be associated with low VWF in other family members. We have recently found a severe form of VWD in which their blood VWF doesn’t bind to exposed collagen after blood vessels are damaged. Current testing as done widely in the United States misses this form of VWD, yet these patients would benefit from VWF replacement therapy. Also we have shown that certain VWF laboratory test (ristocetin-cofactor) is not accurate in as many as 67% of African Americans – they might be assumed to have VWD when they really are normal.
Since our historic diagnosis of VWD might have diagnostic problems, might be develop algorithms that correctly diagnose VWD patients more correctly and provide individualized advice on need for treatment when bleeding is encountered.
For hemophilia we have devised a strategy of gene therapy where we would take a hemophilia patients bone marrow and do gene therapy on his hematopoietic stem cells (blood stem cells) so that the FVIII (or FIX) gene is turned on only in platelets where it is stored in storage granules. If blood vessels are injured the platelet sticks to the site because of VWF and the platelets activate and release the FVIII that was put there by gene therapy and the FVIII accelerates clotting and stops the clinical bleeding. To see if there is any thrombosis risk, we have done similar bone marrow transplants with platelet FVIII into mice with a thrombotic propensity. Adding the platelet FVIII does NOT seem to increase the likelihood of clinical thrombosis. This suggests this approach may be safe.
Our lab has shown that heart valve problems result in the loss of the most active “large” forms of VWF that are most likely to stop clinical bleeding. When infants in their first month of life have major heart surgery for complex congenital heart disease, they sometimes encounter excess abnormal bleeding. Our studies are directed at defining if this bleeding is caused by abnormal VWF and if so, could VWF replacement be optimized to control hemorrhage.
These 5 studies are directed at increasing our understanding of both bleeding and clotting disorders and their appropriate clinical management.